


Introduction
MSFragger is an ultrafast database search tool for peptide identification in mass spectrometry-based proteomics. While we provide a stand-alone Graphical User interface FragPipe for running MSFragger, here we describe the implementation of MSFragger as a processing node in the Thermo Scientific Proteome Discoverer (PD) environment. We also provide PeptideProphet (via Philosopher) as part of the PD processing node, enabling downstream processing of MSFragger search results in PD using either Percolator or PeptideProphet.
The following workflows have been tested and should be fully supported with our MSFragger and PeptideProphet(Philosopher) PD nodes:
- Conventional closed search, label-free or label-based (e.g. TMT)
- Open search
Compared to SEQUEST-HT/Percolator (using a publicly available HEK293 data set PXD001468; conventional closed search), MSFragger/PeptideProphet(Philosopher) reduced the total processing speed by more than a factor of four. For open searches, the improvement in processing speed was even greater. Using MSFragger instead of SEQUEST-HT (with PeptideProphet or Percolator) also resulted in a significant increase in the number of identified proteins/peptides/PSMs.
Page Contents
- Installation
- Requirements
- How to Use
- Processing/Consensus Workflows
- Documentation
- FAQ
- Help/How to Cite
Installation of PD nodes
The MSFragger node can be used with Thermo Scientific Proteome Discoverer versions above 2.2 (not suitable for v2.1 or older versions).
Please follow the steps below to install:
Step 1. Download the latest version of MSFragger-PDv22.zip (or MSFragger-PDv23.zip/MSFragger-PDv24.zip) (depending on your version of Proteome Discoverer) from our github repository and unzip/decompress the file.
Step 2. Make sure that the Proteome Discoverer application is closed.
Step 3. Open the folder where Thermo Scientific Proteome Discoverer is installed. To find this location, right click on your Thermo Scientific Proteome Discoverer desktop icon and then click on “Properties”. The folder path is shown in the field called Target (as shown below).

Step 4. Delete any older versions of MSFragger-PD.dll and copy “MSFragger-PD.dll” from the unzipped file to that folder.
Step 5. Open Thermo Scientific Proteome Discoverer, select the licensing page and click on “Scan for Missing Features”. You should be prompted to close and restart the application.

Step 6. Restart Thermo Scientific Proteome Discoverer and you should see MSFragger and PeptideProphet in your processing nodes (as shown in the Figure).

Step 7. Download the latest versions of MSFragger and Philosopher tools from the corresponding GitHub repositories (they are NOT included with the MSFragger-PD.dll wrapper program). Make sure the MSFragger JAR (.jar extension) file is in the same directory as the ‘ext’ folder, which contains the libraries needed for reading .raw files (i.e., don’t move the .jar file separately from the top MSFragger folder):

MSFragger: Please follow instructions for obtaining the JAR binary file of MSFragger.
Philosopher: You should download the
philosopher_windows_amd64.exefile.
NOTE: the original license agreements for MSFragger and Philosopher also apply when used within the PD environment.
Requirements
-
Java SE Runtime Environment 8 (or above) is required to be installed prior to use MSFragger-PD node.
-
Input files can be RAW, mzML, or mzXML formats.
IMPORTANT: If you choose to use mzML format (instead of RAW), DO NOT use “zlib compression” during file conversion because Proteome Discoverer currently does not support the compression function. An example of parameter setting in file conversion using ProteoWizard/MSconvert is shown in the Figure.

- For searches with multiple PTMs (e.g. phosphorylation) or non-specific digestion searches, MSFragger requires a significant amount of RAM. For such searches, we recommend at least 32Gb of memory, ideally 64Gb or more. For normal tryptic searches, or open searches, even 16Gb should be sufficient. If you would like to perform searches that require significant amount of RAM, we recommend that you use FragPipe instead. FragPipe provides an option for splitting the protein sequence database, circumventing the memory limitations.
How to use
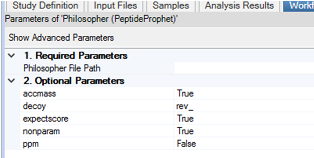
Step 1. Select the binary files of MSFragger and Philosopher in their parameter window.
Step 2. Import your protein fasta file via “Maintain FASTA Files” in PD. Specify the database in MSFragger node. Please note that the fasta file should include only targeted protein sequences. The corresponding decoy sequences will be automatically generated in the MSFragger processing node.
| MSFragger | Philosopher |
|---|---|
 |
 |
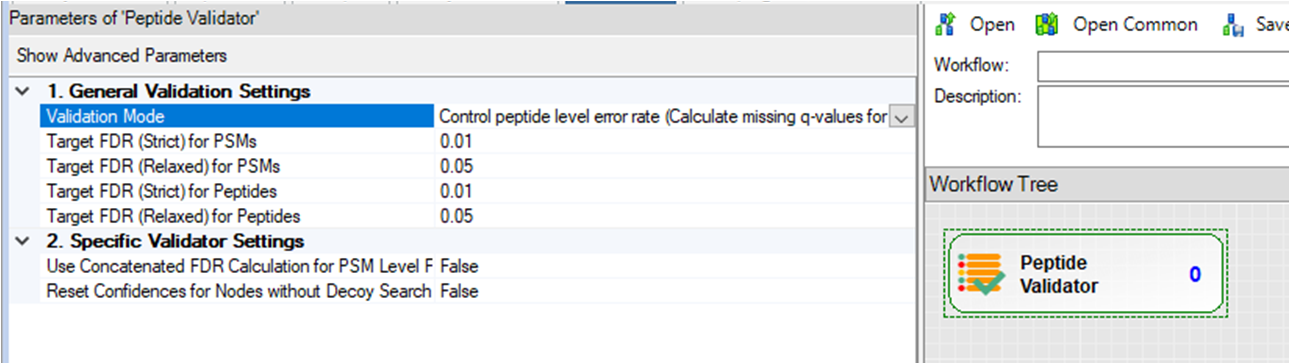
Step 3. When using PeptideProphet, please ensure “Validation Mode=Control peptide level error rate (Calculate missing q-values for PSMs)” in PeptideValidator (Consensus Step) such that the q-values are calculated during the validation.
Step 4. To run multiple processing workflows in parallel (batch mode), please:
(1) set up the value of “Max. Number of Processing Workflows in Parallel Execution” (at Adiministrator -> Configuration -> Parallel Job Execution) as 2 or above (as shown in the figure below).

(2) manually set up the RAM (GB) of MSFragger node in each workflow (as shown in the figure below). A simple way to calculate the RAM is to take the average of available RAMs depending on the number of processing workflows being operated at a time. For example, if you want to run 4 processing workflows at the same time with 64G RAM available, please set up 8GB (=64/4) RAM in the MSFragger node of each workflow. We only recommend running multiple processing workflows for small data sets. If you have a large data set, please set the value of “Max. Number of Processing Workflows in Parallel Execution” to 1, so the processing workflows will be run sequentially.

Processing/Consensus Workflows
We also provide three processing workflows and two consensus workflows for your reference, these can be found in the .zip files you downloaded here.
- Processing_MSFragger_Percolator_ClosedSearch.pdProcessingWF: Use MSFragger and Percolator for conventional closed search.
- Processing_MSFragger_PeptideProphet_ClosedSearch.pdProcessingWF: Use MSFragger and PeptideProphet for conventional closed search.
- Processing_MSFragger_PeptideProphet_OpenSearch.pdProcessingWF: Use MSFragger and PeptideProphet for open search.
- Consensus_Percolator.pdConsensusWF: The consensus workflow for MSFragger and Percolator.
- Consensus_PeptideProphet.pdConsensusWF: The consensus workflor for PeptideProphet.
NOTE: Because of the minor version difference (e.g., PD v2.2.0.385 and PD v2.2.0.388), the workflows sometimes may fail even using the same PD version. The MSFragger parameter files (for closed and open searches) are thus provided along with the workflows for your reference.
Test Data
We recommend the following publicly available HEK293 data set (PXD001468) for testing, since this is what we typically use for testing as well.
Documentation
For documentation on MSFragger itself (hardware requirements, search parameters, etc.), see MSFragger Documentation Wiki page. For Philosopher(PeptideProphet).
Frequently Asked Questions (FAQ)
Q: Why can’t I see the MSFragger PD node after installation?
A: Please check that (1) you have .Net 4.7 or above installed in your computer, (2) the “MSFraggerPDv2x.dll” file is not blocked by your operating system. To unblock it, right click on the file and select ‘Properties’, then in the ‘Attributes’ section, select ‘Advanced’ and then click ‘Unblock’. Alternatively, you can run unblock-file -path "C:\Program Files\Thermo\Proteome Discoverer 2.x\System\Release\MSFraggerPDv2x.dll" (make sure this path is correct) from Windows PowerShell as administrator.
Q: Why I have this error message: Could not create the Java Virtual Machine?
A: Please check if you have the correct version of Java Runtime Environment (JRE) (e.g., 64 and 32 bit) installed in your computer.
Additional Help and Technical Support
Please post all questions/bug reports regarding MSFragger itself on the MSFragger GitHub page, or if more appropriate on Philsopher page.
How to Cite
If you use MSFragger in Proteome Discoverer, we ask that you cite the manuscript describing the PD node (currently in preparation), and well as the manuscripts describing the key individual components:
Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, Nesvizhskii AI. MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nature Methods 14:513–520 (2017). Manuscript.
Leprevost F. et al., Philosopher: a complete toolkit for shotgun proteomics data analysis. Manuscript in preparation.
For other tools developed by Nesvizhskii lab, see our website.